How We Can Classify Organic Chemical Reactions
Classifying Organic Chemical Reactions
This is best accomplished by perceiving the reaction pathway or mechanism of a reaction.
1. Classification by Structural Change
First, we identify four broad classes of reactions based solely on the structural change occurring in the reactant molecules. This classification does not require knowledge or speculation concerning reaction paths or mechanisms.
The letter R in the following illustrations is widely used as a symbol for a generic group. It may stand for simple substituents such as H– or CH3–, or for complex groups composed of many atoms of carbon and other elements.
The letter R in the following illustrations is widely used as a symbol for a generic group. It may stand for simple substituents such as H– or CH3–, or for complex groups composed of many atoms of carbon and other elements.
Four Reaction Classes | ||
|---|---|---|
Addition | Elimination | |
 |  | |
Substitution | Rearrangement | |
 |  | |
In an addition reaction the number of σ-bonds in the substrate molecule increases, usually at the expense of one or more π-bonds. The reverse is true of elimination reactions, i.e.the number of σ-bonds in the substrate decreases, and new π-bonds are often formed. Substitution reactions, as the name implies, are characterized by replacement of an atom or group (Y) by another atom or group (Z). Aside from these groups, the number of bonds does not change. A rearrangement reaction generates an isomer, and again the number of bonds normally does not change.
The examples illustrated above involve simple alkyl and alkene systems, but these reaction types are general for most functional groups, including those incorporating carbon-oxygen double bonds and carbon-nitrogen double and triple bonds. Some common reactions may actually be a combination of reaction types. The reaction of an ester with ammonia to give an amide, as shown below, appears to be a substitution reaction ( Y = CH3O & Z = NH2 ); however, it is actually two reactions, an addition followed by an elimination.
The examples illustrated above involve simple alkyl and alkene systems, but these reaction types are general for most functional groups, including those incorporating carbon-oxygen double bonds and carbon-nitrogen double and triple bonds. Some common reactions may actually be a combination of reaction types. The reaction of an ester with ammonia to give an amide, as shown below, appears to be a substitution reaction ( Y = CH3O & Z = NH2 ); however, it is actually two reactions, an addition followed by an elimination.

The addition of water to a nitrile does not seem to fit any of the above reaction types, but it is simply a slow addition reaction followed by a rapid rearrangement, as shown in the following equation. Rapid rearrangements of this kind are called tautomerizations.

2. Classification by Reaction Type
At the beginning, it is helpful to identify some common reaction types that will surface repeatedly as the chemical behavior of different compounds is examined. This is not intended to be a complete and comprehensive list, but should set the stage for future elaborations.
It is useful to begin a discussion of organic chemical reactions with a review of acid-base chemistry and terminology for several reasons. First, acid-base reactions are among the simplest to recognize and understand. Second, some classes of organic compounds have distinctly acidic properties, and some other classes behave as bases, so we need to identify these aspects of their chemistry. Finally, many organic reactions are catalyzed by acids and/or bases, and although such transformations may seem complex, our understanding of how they occur often begins with the functioning of the catalyst.
Organic chemists use two acid-base theories for interpreting and planning their work: the Brønsted theory and the Lewis theory.Brønsted Theory
According to the Brønsted theory, an acid is a proton donor, and a base is a proton acceptor. In an acid-base reaction, each side of the equilibrium has an acid and a base reactant or product, and these may be neutral species or ions.
| H-A + B:(–) |  | A:(–) + B-H |
| (acid1) (base1) | (base2) (acid2) |
Structurally related acid-base pairs, such as {H-A and A:(–)} or {B:(–) and B-H} are called conjugate pairs. Substances that can serve as both acids and bases, such as water, are termed amphoteric.
| H-Cl + H2O | | Cl:(–) + H3O(+) |
| (acid) (base) | (base) (acid) |
| H3N: + H2O | | NH4(+) + HO(–) |
| (base) (acid) | (acid) (base) |
The relative strength of a group of acids (or bases) may be evaluated by measuring the extent of reaction that each group member undergoes with a common base (or acid). Water serves nicely as the common base or acid for such determinations. Thus, for an acid H-A, its strength is proportional to the extent of its reaction with the base water, which is given by the equilibrium constant Keq.
H-A + H2O | | H3O(+) + A:(–) |  |
Since these studies are generally extrapolated to high dilution, the molar concentration of water (55.5) is constant and may be eliminated from the denominator. The resulting K value is called the acidity constant, Ka. Clearly, strong acids have larger Ka's than do weaker acids. Because of the very large range of acid strengths (greater than 1040), a logarithmic scale of acidity (pKa) is normally employed. Stronger acids have smaller or more negative pKa values than do weaker acids.

Examples of Brønsted Acid-Base Equilibria
| Acid-Base Reaction | Conjugate Acids | Conjugate Bases | Ka | pKa | |||
|---|---|---|---|---|---|---|---|
| HBr + H2O |  | H3O(+) + Br(–) | HBr H3O(+) | Br(–) H2O | 105 | -5 | |
| CH3CO2H + H2O |  | H3O(+) + CH3CO2(–) | CH3CO2H H3O(+) | CH3CO2(–) H2O | 1.77*10-5 | 4.75 | |
| C2H5OH + H2O | | H3O(+) + C2H5O(–) | C2H5OH H3O(+) | C2H5O(–) H2O | 10-16 | 16 | |
| NH3 + H2O | | H3O(+) + NH2(–) | NH3 H3O(+) | NH2(–) H2O | 10-34 | 34 | |
In all the above examples water acts as a common base. The last example ( NH3 ) cannot be measured directly in water, since the strongest base that can exist in this solvent is hydroxide ion. Consequently, the value reported here is extrapolated from measurements in much less acidic solvents, such as acetonitrile.
Since many organic reactions either take place in aqueous environments ( living cells ), or are quenched or worked-up in water, it is important to consider how a conjugate acid-base equilibrium mixture changes with pH. A simple relationship known as the Henderson-Hasselbalch equation provides this information.

When the pH of an aqueous solution or mixture is equal to the pKa of an acidic component, the concentrations of the acid and base conjugate forms must be equal ( the log of 1 is 0 ). If the pH is lowered by two or more units relative to the pKa, the acid concentration will be greater than 99%. On the other hand, if the pH ( relative to pKa ) is raised by two or more units the conjugate base concentration will be over 99%. Consequently, mixtures of acidic and non-acidic compounds are easily separated by adjusting the pH of the water component in a two phase solvent extraction.
For example, if a solution of benzoic acid ( pKa = 4.2 ) in benzyl alcohol ( pKa = 15 ) is dissolved in ether and shaken with an excess of 0.1 N sodium hydroxide ( pH = 13 ), the acid is completely converted to its water soluble ( ether insoluble ) sodium salt, while the alcohol is unaffected. The ether solution of the alcohol may then be separated from the water layer, and pure alcohol recovered by distillation of the volatile ether solvent. The pH of the water solution of sodium benzoate may then be lowered to 1.0 by addition of hydrochloric acid, at which point pure benzoic acid crystallizes, and may be isolated by filtration.
For example, if a solution of benzoic acid ( pKa = 4.2 ) in benzyl alcohol ( pKa = 15 ) is dissolved in ether and shaken with an excess of 0.1 N sodium hydroxide ( pH = 13 ), the acid is completely converted to its water soluble ( ether insoluble ) sodium salt, while the alcohol is unaffected. The ether solution of the alcohol may then be separated from the water layer, and pure alcohol recovered by distillation of the volatile ether solvent. The pH of the water solution of sodium benzoate may then be lowered to 1.0 by addition of hydrochloric acid, at which point pure benzoic acid crystallizes, and may be isolated by filtration.
Basicity
The basicity of oxygen, nitrogen, sulfur and phosphorus compounds or ions may be treated in an analogous fashion. Thus, we may write base-acid equilibria, which define a Kb and a corresponding pKb. However, a more common procedure is to report the acidities of the conjugate acids of the bases ( these conjugate acids are often "onium" cations ). The pKa's reported for bases in this system are proportional to the base strength of the base. A useful rule here is: pKa + pKb = 14.
We see this relationship in the following two equilibria:
We see this relationship in the following two equilibria:
| Acid-Base Reaction | Conjugate Acids | Conjugate Bases | K | pK | |||
|---|---|---|---|---|---|---|---|
| NH3 + H2O | | NH4(+) + OH(–) | NH4(+) H2O | NH3 OH(–) | Kb = 1.8*10-5 | pKb = 4.74 | |
| NH4(+) + H2O | | H3O(+) + NH3 | NH4(+) H3O(+) | NH3 H2O | Ka = 5.5*10-10 | pKa = 9.25 | |
Tables of pKa values for inorganic and organic acids ( and bases) are available in many reference books, and may be examined here by clicking on the appropriate link:
Although it is convenient and informative to express pKa values for a common solvent system (usually water), there are serious limitations for very strong and very weak acids. Thus acids that are stronger than the hydronium cation, H3O(+), and weak acids having conjugate bases stronger than hydroxide anion, OH(–), cannot be measured directly in water solution. Solvents such as acetic acid, acetonitrile and nitromethane are often used for studying very strong acids. Relative acidity measurements in these solvents may be extrapolated to water. Likewise, very weakly acidic solvents such as DMSO, acetonitrile, toluene, amines and ammonia may be used to study the acidities of very weak acids. For both these groups, the reported pKa values extrapolated to water are approximate, and many have large uncertainties.
Lewis Theory
According to the Lewis theory, an acid is an electron pair acceptor, and a base is an electron pair donor. Lewis bases are also Brønsted bases; however, many Lewis acids, such as BF3, AlCl3 and Mg2+, are not Brønsted acids. The product of a Lewis acid-base reaction, is a neutral, dipolar or charged complex, which may be a stable covalent molecule. As shown at the top of the following drawing, coordinate covalent bonding of a phosphorous Lewis base to a boron Lewis acid creates a complex in which the formal charge of boron is negative and that of phosphorous is positive. In this complex, boron acquires a neon valence shell configuration and phosphorous an argon configuration. If the substituents (R) on these atoms are not large, the complex will be favored at equilibrium. However, steric hindrance of bulky substituents may prohibit complex formation. The resulting mixture of non-bonded Lewis acid/base pairs has been termed "frustrated", and exhibits unusual chemical behavior.
Two examples of Lewis acid-base equilibria that play a role in chemical reactions are shown in equations 1 & 2 below.
Two examples of Lewis acid-base equilibria that play a role in chemical reactions are shown in equations 1 & 2 below.

In the first example, an electron deficient aluminum atom bonds to a covalent chlorine atom by sharing one of its non-bonding valence electron pairs, and thus achieves an argon-like valence shell octet. Because this sharing is unilateral (chlorine contributes both electrons), both the aluminum and the chlorine have formal charges, as shown. If the carbon chlorine bond in this complex breaks with both the bonding electrons remaining with the more electronegative atom (chlorine), the carbon assumes a positive charge. We refer to such carbon species as carbocations. Carbocations are also Lewis acids, as the reverse reaction demonstrates.
Many carbocations (but not all) may also function as Brønsted acids. Equation 3 illustrates this dual behavior; the Lewis acidic site is colored red and three of the nine acidic hydrogen atoms are colored orange. In its Brønsted acid role the carbocation donates a proton to the base (hydroxide anion), and is converted to a stable neutral molecule having a carbon-carbon double bond.
Many carbocations (but not all) may also function as Brønsted acids. Equation 3 illustrates this dual behavior; the Lewis acidic site is colored red and three of the nine acidic hydrogen atoms are colored orange. In its Brønsted acid role the carbocation donates a proton to the base (hydroxide anion), and is converted to a stable neutral molecule having a carbon-carbon double bond.

A terminology related to the Lewis acid-base nomenclature is often used by organic chemists. Here the term electrophile corresponds to a Lewis acid, and nucleophile corresponds to a Lewis base.
Electrophile: An electron deficient atom, ion or molecule that has an affinity for an electron pair, and will bond to a base or nucleophile.
Nucleophile: An atom, ion or molecule that has an electron pair that may be donated in bonding to an electrophile (or Lewis acid).
Electrophile: An electron deficient atom, ion or molecule that has an affinity for an electron pair, and will bond to a base or nucleophile.
Nucleophile: An atom, ion or molecule that has an electron pair that may be donated in bonding to an electrophile (or Lewis acid).
Oxidation and Reduction Reactions
A parallel and independent method of characterizing organic reactions is by oxidation-reduction terminology. Carbon atoms may have any oxidation state from –4 (e.g. CH4 ) to +4 (e.g. CO2 ), depending upon their substituents. Fortunately, we need not determine the absolute oxidation state of each carbon atom in a molecule, but only the change in oxidation state of those carbons involved in a chemical transformation. To determine whether a carbon atom has undergone a redox change during a reaction we simply note any changes in the number of bonds to hydrogen and the number of bonds to more electronegative atoms such as O, N, F, Cl, Br, I, & S that has occurred. Bonds to other carbon atoms are ignored. This count should be conducted for each carbon atom undergoing any change during a reaction.
- If the number of hydrogen atoms bonded to a carbon increases, and/or if the number of bonds to more electronegative atoms decreases, the carbon in question has been reduced (i.e. it is in a lower oxidation state).
- If the number of hydrogen atoms bonded to a carbon decreases, and/or if the number of bonds to more electronegative atoms increases, the carbon in question has been oxidized (i.e. it is in a higher oxidation state).
- If there has been no change in the number of such bonds, then the carbon in question has not changed its oxidation state. In the hydrolysis reaction of a nitrile shown above, the blue colored carbon has not changed its oxidation state.
These rules are illustrated by the following four addition reactions involving the same starting material, cyclohexene. Carbon atoms colored blue are reduced, and those colored red are oxidized. In the addition of hydrogen both carbon atoms are reduced, and the overall reaction is termed a reduction. Peracid epoxidation and addition of bromine oxidize both carbon atoms, so these are termed oxidation reactions. Addition of HBr reduces one of the double bond carbon atoms and oxidizes the other; consequently, there is no overall redox change in the substrate molecule.

Since metals such as lithium and magnesium are less electronegative than hydrogen, their covalent bonds to carbon are polarized so that the carbon is negative (reduced) and the metal is positive (oxidized). Thus, Grignard reagent formation from an alkyl halide reduces the substituted carbon atom. In the following equation and half-reactions the carbon atom (blue) is reduced and the magnesium (magenta) is oxidized.
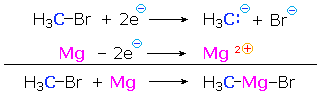
3. Classification by Functional Group
Functional groups
Are atoms or small groups of atoms (usually two to four) that exhibit a characteristic reactivity when treated with certain reagents. To view a table of the common functional groups and their class names Click Here. A particular functional group will almost always display its characteristic chemical behavior when it is present in a compound. Because of this, the discussion of organic reactions is often organized according to functional groups. The following table summarizes the general chemical behavior of the common functional groups. For reference, the alkanes provide a background of behavior in the absence of more localized functional groups.
Are atoms or small groups of atoms (usually two to four) that exhibit a characteristic reactivity when treated with certain reagents. To view a table of the common functional groups and their class names Click Here. A particular functional group will almost always display its characteristic chemical behavior when it is present in a compound. Because of this, the discussion of organic reactions is often organized according to functional groups. The following table summarizes the general chemical behavior of the common functional groups. For reference, the alkanes provide a background of behavior in the absence of more localized functional groups.
| Functional Class | Formula | Characteristic Reactions |
|---|---|---|
| Alkanes | C–C, C–H | Substitution (of H, commonly by Cl or Br) Combustion (conversion to CO2 & H2O) |
| Alkenes | C=C–C–H | Addition Substitution (of H) |
| Alkynes | C≡C–H | Addition Substitution (of H) |
| Alkyl Halides | H–C–C–X | Substitution (of X) Elimination (of HX) |
| Alcohols | H–C–C–O–H | Substitution (of H); Substitution (of OH) Elimination (of HOH); Oxidation (elimination of 2H) |
| Ethers | (α)C–O–R | Substitution (of OR); Substitution (of α–H) |
| Amines | C–NRH | Substitution (of H); Addition (to N); Oxidation (of N) |
| Benzene Ring | C6H6 | Substitution (of H) |
| Aldehydes | (α)C–CH=O | Addition Substitution (of H or α–H) |
| Ketones | (α)C–CR=O | Addition Substitution (of α–H) |
| Carboxylic Acids | (α)C–CO2H | Substitution (of H); Substitution (of OH) Substitution (of α–H); Addition (to C=O) |
| Carboxylic Derivatives | (α)C–CZ=O (Z = OR, Cl, NHR, etc.) | Substitution (of Z); Substitution (of α–H) Addition (to C=O) |
This table does not include any reference to rearrangement, due to the fact that such reactions are found in all functional classes, and are highly dependent on the structure of the reactant. Furthermore, a review of the overall reaction patterns presented in this table discloses only a broad and rather non-specific set of reactivity trends. This is not surprising, since the three remaining categories provide only a coarse discrimination (comparable to identifying an object as animal, vegetable or mineral). Consequently, apparent similarities may fail to reflect important differences. For example, addition reactions to C=C are significantly different from additions to C=O, and substitution reactions of C-X proceed in very different ways, depending on the hybridization state of carbon.

Comments